

The Medical Devices field is a constantly moving area. Several devices are invented or improved every day to benefit patients. The new Regulation (EU) 2017/745, which is more demanding for the manufacturers, aims to improve device safety while supporting innovation on the European Market. Post Market Surveillance (PMS) and Post-market Clinical Follow-up (PMCF) whose plan is an integral part of the PMS plan.
The PMCF (Post-Market Clinical Follow-up) is carried out following the release of a medical device onto the market. Following the device’s CE marking, the PMCF aims to be able to update the clinical evaluation report with updated data on patient safety and device performance.
The PMCF is defined as “a continuous process that updates the clinical evaluation referred to in Article 61 and Part A of this Annex and shall be addressed in the manufacturer’s post-market surveillance plan” in Annex XIV Part B of the MDR, which is entirely dedicated to it”. These activities can include anything from conducting a new clinical trial to analyzing data from already registered devices. Medical devices manufacturers are advised by European and American regulatory authorities to include PMCF in their Post Market Surveillance Plan since clinical data collected during the pre-market period may be too limited to identify rare events or incident. Post Market Clinical Follow-up is crucial to identify new and unknown risks.
A PMCF plan should outline the overall PMCF activities, and a PMCF report that is a part of the device’s Clinical Evaluation Report (CER) should compile the data.
PMS Plan
- Details the strategy for continously monitoring and collecting information avout safety and performance of a device throughout its entire lifetime
- Incorporates PMCF activities and vigilance systems
- Is part of the manufacturer’s quality system
PMCF Plan
- Outlines the manufacturer’s planned procedures and methods for collecting clinical data for post-market follow-up
- Complements the data obtained during the pre-market phase
PMS Report
- Is produced to provide the results obtained during PMCF activities
- Is fully integrated as part of the technical documentation and more specifically, it’s outcome included in the clinical evaluation report
CER
- Gathers the clinical evidence and documents the conclusions of a clinical evaluation of a medical device
- Is an essential step in obtaining CE marking of a device and must be regulary updated during the PMS
PMS Plan System
The MEDDEV documents were not legally binding but were summarizing the consensus of various experts, used as guidelines, and created for the MDD. Even though, in general, the MEDDEV 2.7.1 rev. 4 on the clinical evaluation is partly inspired by the Medical device Regulation (MDR), it remains insufficient to presume compliance with the requirements of the MDR. The PMS system has now been more defined and fully integrated within the Regulation (EU) 2017/745.
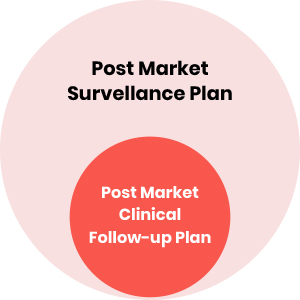
As part of the Post-Market Surveillance system, the Post Market Clinical Follow-up activities are considered by the MDR as a continuous process that updates the clinical evaluation, and, as described above, the PMCF plan is an integral part of the PMS plan.
Scope of PMCF
The document intends to offer advice on the creation, execution, and suitable use of PMCF studies.
It provides guidance in relation to:
- The circumstances where a PMCF study is indicated
- The objectives of PMCF Studies
- The design and implementation of PMCF studies
- The use of information from PMCF studies
The aim of the PMCF plan is as follows:
- Confirming the safety and performance, including the clinical benefit if applicable, of the device throughout its expected lifetime
- Identifying previously unknown side-effects and monitor the identified side-effects and contraindications
- Identifying and analyzing emergent risks on the basis of factual evidence
- Ensuring the continued acceptability of the benefit-risk ratio, referred to in section 1 and 9 of Annex I in the MDR
- Identifying possible systematic misuse or off-label use of the device, with a view to verifying that the intended purpose is correct
The circumstances where a PMCF study is indicated:
- PMCF studies can be done to gather further clinical information to resolve any unanswered questions concerning a device
- PMCF studies might also be necessary to address new challenges issues brought about by post market adverse event patterns, data from the literature, signals from adverse event reports, active surveillance programmes, or information from other sources
Objective of PMCF
- Identifying and investigating residual risks associated with use of the device
- Contributing towards the update of Clinical Evaluation
- Detecting any emerging risks and previously unknown side-effects
- Confirming the overall safety and performance of the medical device in normal use
- Identifying systematic misuse of the device and its impact on safety and performance
PMCF investigations are carried out to monitor the long-term effects of the medical devices and any probable risks or complications that may arise. It also helps to identify any potential risks associated with the use of the product and to identify any trends or patterns in adverse events associated with its use. The results of these investigations can be used to inform regulatory decisions related to the product’s safety and efficacy.
PMCF Activities
Performance
Specific requirements for designing, conducting and reporting clinical investigations are documented in Annex XV of the MDR.
First, PMCF studies must comply with applicable laws and regulations, and follow relevant guidelines and standards.
The design and methodology of the PMCF study must be described in a Clinical Investigation Plan (CIP) or study protocol that will include a description of the rationale, objectives, endpoints, and statistical methods that must be adapted to achieve the stated objectives. The study must be carried out with adequate controls in place to be in full compliance with the CIP.
A Statistical Analysis Plan (SAP) is also required to provide a detailed description of the statistical methods. It is this document that will dictate how the analysis of the data is to be performed by a person with appropriate statistical expertise.
The final Study Report should describe the results of the analysis in detail and provide conclusions related to the intended objectives.
Design considerations for PMCF under the MDR
Manufacturers should take a strategic approach when creating a PMCF system to satisfy MDR criteria, which frequently entails using specialized types of clinical research to satisfy the whole range of standards. In order to comply with PMCF standards for longitudinal data collection over the course of a device’s lifetime, the majority of “standard” clinical trials only collect data for a short period of time and on a small population of patients.
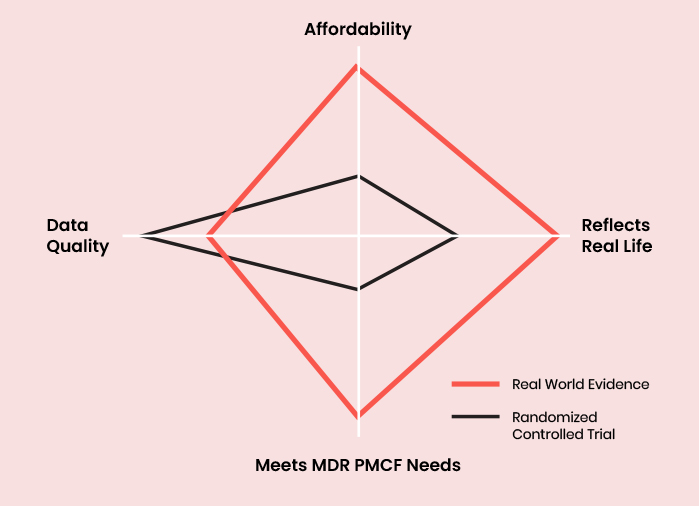
PMCF systems under the MDR should be designed to produce Real World Evidence (RWE) on device performance in normal use. RWE is well-suited to meeting the requirements of MDR PMCF because it:
- Captures data from a study / survey population that represents the entire population normally exposed to the device
- Is non-comparative, focusing only on the safety and performance of the subject device rather than making comparisons between different devices
- Does not involve experimental exposure; rather, it studies the use of a device that is already CE-marked and seeks to confirm or refute that it meets the necessary safety and performance requirements
- Recruits an unlimited number of patients over time and runs indefinitely
Is conducted at all types of clinical site and does not select for centres of excellence of other sites that may be unrepresentative or positively biased towards better results
A Comparison Between Real World Evidence and Standard Clinical Data From A Randomised Control Trial
The Notified body shall always decide based on the result of its assessment of the clinical evaluation and risk management whether the post market surveillance plan, including the PMCF plan is adequate.
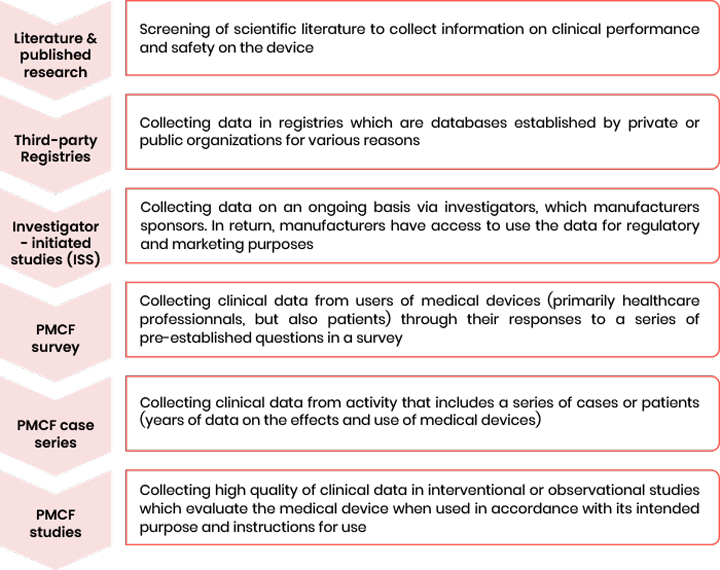
A PMCF can follow several methodologies such as:
- The extend follow up of patient enrolled in premarket investigation
- A new clinical investigation
- A review of data derived from a device registry
- A review of relevant retrospective data from patients previously exposed to the device
For CE marking application of medical device, all medical devices are required to have evidence of PMCF study protocol or a justification for why a post market clinical follow up study is not required.
There are new risk that are identified when the device is used for a long duration (e.g implantable) by a broader patient population because the medium/long term safety and clinical performance are already known from previous use of the device and other appropriate post market surveillance activities have provide sufficient data to address the risk.
Tests are rarely adequate for determining an implant’s longevity, usability by all intended users, and patient satisfaction. If tests and pre-market clinical studies are insufficient, a post-market follow-up study must be conducted to address the specific residual risk.
In addition, clinical data/evidence generated from PMCF studies can be used to:
- Become part of pre-market clinical evidence, or supplementary data for next generation or similar technologies when applying for marketing authorization
- Develop objective performance criteria and performance goals
- Form control/comparison groups
Conclusion
The conclusions of the PMCF evaluation report shall be taken into account for the clinical evaluation referred to in Article 61 and Part A of this Annex and in the risk management referred to in Section 3 of Annex I. If, through the PMCF, the need for preventive and/or corrective measures has been identified, the manufacturer shall implement them. This may result in the need to reassess whether the device continues to comply with the Essential Principles. Such assessment may result in corrective or preventive actions, for example:
- Changes to the labelling/instructions for use
- Changes to manufacturing processes
- Changes to the device design
- Public health notifications, or
- Product removal
It is always advantageous to have a methodical approach to report design. MakroCare, with more than 15 years of experience, has optimized these processes to ensure quality and success for manufacturers.
Let MakroCare be your partner in overcoming post-market surveillance challenges.
